El software, desarrollado por científicos de Argentina y de España, identifica mutaciones genéticas asociadas a tumores por lo que servirá para evitar el uso innecesario de terapias costosas y con efectos secundarios en pacientes que no se verán beneficiados.
(Agencia CyTA-Leloir. Por Bruno Geller)-. Una de las terapias más avanzadas contra el cáncer, la inmunoterapia, aumenta la supervivencia de pacientes con cáncer de pulmón, melanoma, carcinoma de células renales, linfoma de Hodgkin y otros tumores. Sin embargo, no todos los pacientes tienen una respuesta satisfactoria a este tratamiento.
Ahora, investigadores de Argentina y de España pusieron a punto un método bioinformático para mejorar la selección de pacientes que responderán a la inmunoterapia para el tratamiento de distintos tumores.
“Desarrollamos modelos matemáticos, que integrados en un software, dan una medida muy precisa de la carga mutacional de pacientes para 14 tumores diferentes. Nuestra herramienta puede agilizar y mejorar la selección de pacientes que se verían beneficiados con inmunoterapia”, señaló Cristina Marino-Buslje, directora del estudio, jefa del Laboratorio de Bioinformática Estructural de la FIL e investigadora del CONICET.
Aplicar inmunoterapias disponibles en pacientes que no se verán beneficiados resulta un perjuicio dado los efectos secundarios de la terapia y el costo del tratamiento. Por lo tanto, encontrar un método que a priori indique quienes se beneficiarán de la terapia con mayor probabilidad, es una tarea que ha ocupado a los científicos los últimos años. Al momento se aplican costosas y no tan precisas herramientas de análisis genético que intentan predecir quienes se verían beneficiados.
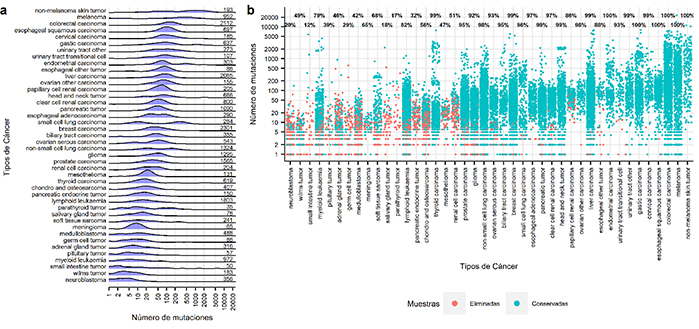
Pero recientemente se ha visto que la cantidad de mutaciones (cambios genéticos) que posee el tumor del paciente está correlacionada con la respuesta que tendrá a la terapia inmunológica. La cantidad de mutaciones totales del tumor o TMB, por las siglas en inglés, se realiza secuenciando todo el genoma o exoma (fracción del ADN que codifica la fabricación de proteínas), pero es un método laborioso, costoso y de difícil aplicación clínica.
Para dar solución a este problema y hacer más factible este análisis, se utilizan los llamados paneles de genes, es decir se secuencian solo un conjunto de genes, aunque los que están disponibles comercialmente “dan una medida poco precisa y en ciertos casos inútil”, explicó Marino-Buslje.
Para contribuir a la solución de este problema, los investigadores desarrollaron un método bioinformático predictivo de la respuesta a la inmunoterapia para 14 tipos de tumores que incluyen las neoplasias malignas sólidas más comunes, incluyendo cánceres de pulmón, colon, piel (melanoma), próstata y mama.
“El método consiste en contar el número de mutaciones en unos pocos genes específicos (que deberán ser secuenciados) de cada tipo de tumor y, con ese número, a través de un modelo matemático, inferir la cantidad total de mutaciones que tiene el individuo”, señaló Miguel Ángel Molina-Vila, también director del estudio e investigador del Hospital Universitario Quirón Dexeus, en Barcelona, España.
“Los paneles de genes disponibles en el mercado están diseñados para dar otras respuestas, son inadecuados para calcular la TMB y esto puede derivar en la clasificación errónea de hasta un tercio de los pacientes. Esto sucede porque se usa un solo panel (lista de genes a analizar) para todos los tipos de cáncer, sin tener en cuenta que cada tumor tiene distintos genes mutados”, destacó Elizabeth Martínez Pérez, primera autora del estudio e investigadora del CONICET en la FIL. “Por esta razón, decidimos desarrollar un software que facilite una mejor predicción de TMB para cada tipo de tumor”, añadió.
Analizando miles de muestras de pacientes con cáncer disponibles en bases públicas de datos de todo el mundo, Marino-Buslje, Molina-Vila y Pérez Martínez lograron captar cuáles serían los paneles óptimos para cada tipo de tumor y hacer un modelo matemático que logre una predicción precisa de la carga mutacional total para cada uno de los 14 tipos de cáncer.
Los paneles y software propuestos por los autores fueron creados analizando casi 25 mil muestras y validados con datos externos provenientes de artículos publicados y bases de datos internacionales como el Atlas del Genoma del Cáncer (TCGA), el Consorcio Internacional del Genoma del Cáncer (ICGC), el Proyecto Genoma del Cáncer (CGP) y el Catálogo de Mutaciones Somáticas en Cáncer (COSMIC). Los resultados del estudio fueron publicados en la revista “NPJ Precision Oncology”.
“Estamos contentos ya que nuestro trabajo puede facilitar una predicción más precisa de la TMB y así discriminar qué pacientes van a responder a las inmunoterapias en esos tipos de cáncer. Además, esos resultados se logran secuenciando unos pocos genes. Dos ventajas importantes en comparación con los paneles actuales que incluyen más genes incrementando el costo y tienen menor desempeño, conduciendo en algunos casos a una decisión errónea respecto a una terapia. El uso innecesario de terapias en pacientes que no se beneficiarán de ella es perjudicial ya que traen aparejados algunos efectos secundarios y son muy costosas”, indicó Molina-Vila.
Ahora los investigadores están desarrollando una página web de acceso libre y gratuito y fácil uso, que permitirá el cálculo de la TMB a partir del número de mutaciones de los genes indicados para cada tumor. “Esperamos tener una interfase de muy fácil uso. Nuestra idea es que sea una herramienta de uso diario entre los profesionales de la salud, que colabore a identificar qué pacientes responderían bien a las inmunoterapias hoy disponibles en medicina”, indicaron.
Si bien el estudio incluyó una gran cantidad de muestras (más de 24 mil), los investigadores afirmaron que son pocas comparadas con la incidencia mundial del cáncer (19 millones de personas en 2020). “Cuantos más datos tengamos para procesar, mejores, más precisos y más representativos serán los resultados. Quisiéramos exhortar a la comunidad médica de oncología y análisis genómicos a que hagan públicos en estas bases de datos internacionales los análisis genómicos de los pacientes (siguiendo las pautas de ética, los pacientes siempre serán anónimos y no identificables) y así contar con mayor cantidad de datos para hacer ciencia de alta calidad”, subrayaron.

Los autores del estudio: Cristina Marino-Buslje (izq.), Miguel Anguel Molina-Vila y Elizabeth Pérez Martínez.
El software brinda una medida muy precisa de la carga mutacional de pacientes para 14 tumores diferentes. La información será útil para mejorar la selección de quienes responderán a la inmunoterapia.